Press Release.

BOSTON and LONDON, April 22, 2024 (GLOBE NEWSWIRE) — Centessa Pharmaceuticals plc (Nasdaq: CNTA), a clinical-stage pharmaceutical company that aims to discover and develop medicines that are transformational for patients, today announced that the U.S. Food and Drug Administration (FDA) has cleared the Investigational New Drug application (IND) to initiate a Phase 1 first-in-human, clinical trial of ORX750 for the treatment of narcolepsy. ORX750 is an investigational, orally administered, highly potent and selective orexin receptor 2 (OX2R) agonist designed to directly target the underlying pathophysiology of orexin neuron loss in narcolepsy type 1 (NT1), with potential applicability to narcolepsy type 2 (NT2), idiopathic hypersomnia (IH), and other sleep-wake disorders with normal orexin levels.
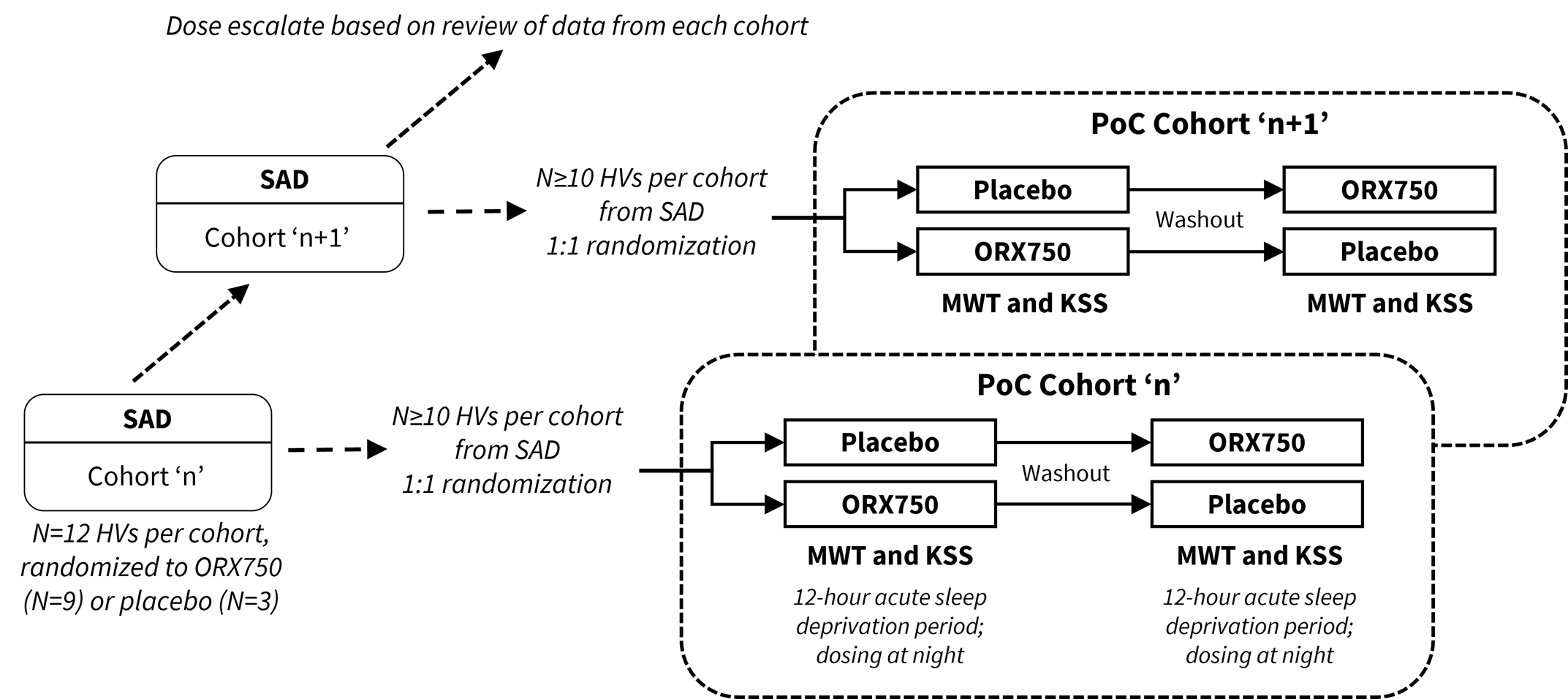
The Phase 1 study will evaluate the safety, tolerability and pharmacokinetics of single-ascending doses (SAD) and multiple-ascending doses (MAD) of ORX750 in healthy adult subjects. In parallel to the SAD, a cross-over pharmacodynamic (PD) assessment will be performed utilizing the Maintenance of Wakefulness Test (MWT) and Karolinska Sleepiness Scale (KSS) in acutely sleep-deprived healthy adult subjects which is intended to provide proof-of-concept data to enable dose selection for NT1, NT2 and IH indications. The study has a maximum exposure limit specified by the FDA which the Company believes significantly exceeds the predicted efficacious doses of ORX750 in indications associated with or without orexin loss; therefore, the Company does not expect this limit to affect any of the planned clinical development activities for ORX750. The Company expects to commence dosing of the Phase 1 study in healthy volunteers imminently, and proof-of-concept data are anticipated in the second half of 2024.
The Phase 1 study design of ORX750 includes SAD combined with PoC cohorts to assess PD effects of ORX750 by measuring sleep latency with the MWT and subjective sleepiness with the KSS in acutely sleep-deprived healthy subjects.
“This is a significant milestone for the development of our potential best-in-class OX2R agonist, ORX750, for the treatment of narcolepsy and other sleep-wake disorders,” said Saurabh Saha MD PhD, Chief Executive Officer of Centessa. “We are excited to begin executing what we believe is an elegant, adaptive Phase 1 study aimed at generating early proof-of-concept data for ORX750 in acutely sleep-deprived healthy volunteers in the second half of this year. We expect this study to enable dose selection for planned studies evaluating ORX750 in patients with NT1 and in patient populations with normal orexin levels, including NT2 and IH.”
About ORX750
ORX750 is an investigational, orally administered, highly potent and selective orexin receptor 2 (OX2R) agonist designed to directly target the underlying pathophysiology of orexin neuron loss in narcolepsy type 1 (NT1). ORX750 has been shown to potently activate the OX2R with an in vitro EC50 of 0.11 nM and 9,800-fold selectivity over the human orexin receptor (hOX1R). ORX750 is Centessa’s first orexin product candidate being developed for the treatment of narcolepsy with potential expansion into narcolepsy type 2 (NT2), idiopathic hypersomnia (IH) and other sleep-wake disorders.
About Centessa Pharmaceuticals
Centessa Pharmaceuticals plc is a clinical-stage pharmaceutical company that aims to discover and develop transformational medicines for patients. Our most advanced programs include a hemophilia program, an orexin agonist program for the treatment of narcolepsy and other sleep-wake disorders and an immuno-oncology program focused on our LockBody® technology platform. We operate with the conviction that each of our programs has the potential to change the current treatment paradigm and establish a new standard of care. For more information, visit www.centessa.com, which does not form part of this release.
Forward Looking Statements
This press release contains forward-looking statements. These statements may be identified by words such as “may,” “might,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “objective,” “anticipate,” “believe,” “estimate,” “predict,” “potential,” “continue,” “ongoing,” “aim,” “seek,” and variations of these words or similar expressions that are intended to identify forward-looking statements. Any such statements in this press release that are not statements of historical fact may be deemed to be forward-looking statements, including statements related to the Company’s ability to discover and develop transformational medicines for patients; the timing of commencement of new studies or clinical trials of ORX750 and other potential orexin agonist candidates; research and clinical development plans and the timing thereof; the Company’s ability to differentiate ORX750 and other potential orexin agonist candidates from other treatment options; the development and therapeutic potential of ORX750 and other potential orexin agonist candidates; predicted efficacious doses of ORX750; the Company’s ability to successfully conduct its clinical development of ORX750 below the maximum exposure limit set by the FDA or, in the event the Company plans to exceed the maximum exposure limit, the Company’s ability to successfully have the maximum exposure limit removed; and other regulatory matters, including the timing and likelihood of success of obtaining authorizations to initiate or continue clinical trials. Any forward-looking statements in this press release are based on our current expectations, estimates and projections only as of the date of this release and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, risks related to the safety and tolerability profile of our product candidates; our ability to protect and maintain our intellectual property position; business (including commercial viability), regulatory, economic and competitive risks, uncertainties, contingencies and assumptions about the Company; risks inherent in developing product candidates and technologies; future results from our ongoing and planned clinical trials; our ability to obtain adequate financing, including through our financing facility with Oberland, to fund our planned clinical trials and other expenses; trends in the industry; the legal and regulatory framework for the industry, including the receipt and maintenance of clearances to conduct or continue clinical testing; future expenditures risks related to our asset-centric corporate model; the risk that any one or more of our product candidates will not be successfully developed and/or commercialized; the risk that the results of preclinical studies or clinical studies will not be predictive of future results in connection with future studies; geo-political risks such as the Russia-Ukraine war and conflicts in the Middle East. These and other risks concerning our programs and operations are described in additional detail in our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, and our other reports, which are on file with the U.S. Securities and Exchange Commission (SEC). We explicitly disclaim any obligation to update any forward-looking statements except to the extent required by law.
Contact:
Kristen K. Sheppard, Esq.
SVP of Investor Relations
investors@centessa.com
A photo accompanying this announcement is available at https://www.globenewswire.com/NewsRoom/AttachmentNg/b64204a9-7c64-47fd-b834-598ca0e45978









